Ферменты являются наиболее эффективными и специфичными из всех известных катализаторов. Каталитические свойства ферментов локализованы в их активных центрах, которые образованы относительно небольшим количеством аминокислотных остатков. Функция остальной части полипептидной цепи состоит в том, что она удерживает активные центры в пространственном положении, необходимом для осуществления специфической каталитической функции.
Во многих случаях для катализа ферментативных систем необходимы, кроме субстрата и фермента, вещества небелковой природы — кофакторы. Последними могут служить либо органические соединения (коферменты), либо ионы металлов.
Часто коферментами являются витамины.
Взаимодействие ионов металлов с ферментами в химическом отношении является частным проявлением более общей закономерности— образования металлоорганических комплексов, основного типа соединений в биологических системах. Комплексообразование в большей или меньшей степени свойственно всем элементам периодической системы, однако наиболее выражена эта способность у катионов металлов.
В комплексном соединении ион металла занимает центральное место. Ионы или молекулы, образующие комплекс с металлом, называются аддендами или лигандами. Простыми анионными лигандами in vivo являются СО32-, НСО32-, РО43-, HPO42-, Н2РО4—, SO42-, F—, Cl—, Br—, J—, вода. Наряду с этим лигандами может служить большое число природных органических соединений. Это аминокислоты, пептиды, белки, нуклеопротеиды, нуклеиновые кислоты, карбоновые кислоты, углеводы, фосфолипиды и другие соединения, содержащие электронно-донорные атомы (азот, кислород, серу).
Предпочтение, отдаваемое ионом металла той или иной группе, зависит от ряда факторов: характера металла, природы лиганда, особенности растворителя и др. Прочность образуемых комплексов ион металла-лиганд хорошо объясняется с позиции концепции жестких и мягких кислот и оснований.
Согласно электронной теории кислот и оснований Льюиса, акцепторы электронов (атомы, ионы или молекулы) играют роль кислот, а доноры электронов — оснований.
Кислоты или основания делятся, в свою очередь, на жесткие и мягкие. Мягкость или жесткость обусловлены подвижностью электронов, или поляризуемостью частиц. При легком смещении электронов частицы относят к мягким, при трудном — к жестким. Катионы щелочных, щелочноземельных и переходных биоэлементов-металлов являются в основном жесткими кислотами, а неорганические анионы — жесткими основаниями. Прочные связи образуют только жесткие кислоты с жесткими основаниями и мягкие кислоты с мягкими основаниями.
Связь между противоположными по жесткости кислотами и основаниями всегда будет слабой, если она вообще возможна.
Стабильность комплекса ион двухвалентного металла-лиганд (зависящий от порядка изменения мягкости) изменяется согласно ряду Ирвинга-Уильямса следующим образом:
Ca < Mg < Mn < Fe < Co << Ni < Cu > Zn,
т. е. соответствует переходу от жесткой кислоты Са2+ к иону с промежуточной жесткостью Cu2+.
Мягкость лигандов, или их способность быть донорами электронов, также влияет на устойчивость комплекса и селективность ионов металла. Так, у белков при переходе от кислородных электронно-донорных функциональных групп (жестких) к сернистым (мягким) меняется порядок предпочтения металла.
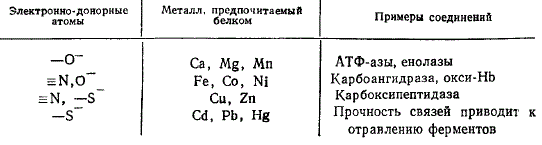
Соответствие между донорными атомами в белках и ионами металлов
При взаимодействии некоторых ионов с органическими соединениями (аминокислотами, пептидами, белками) происходит процесс хелатирования, т. е. образования клешневидных комплексов.
О хелатировании говорят, когда ион металла связывается с двумя или более атомами одного лиганда.
За комплексирование в этих группах ответственны атомы N, О, S. Эти элементы располагают электронными парами, с помощью которых осуществляется координационная связь и заполнение электронной конфигурации центрального иона.
Особо прочные комплексы образуются при наличии реакционноспособных групп с отрицательным зарядом в боковых цепях аминокислот. Присутствие таких групп в гистидине, цистине, цистеине делает их наиболее сильными комплексонами среди аминокислот. Комплексы ионов с пептидами менее прочны, чем с аминокислотами.
Координационные соединения (хелаты) — это наиболее выгодная для организма форма взаимодействия металла с лигандом. Активность элементов в этих комплексах возрастает часто в сотни тысяч и миллионы раз в сравнении с активностью металла в ионном состоянии.
Понятие «координационные соединения» более узкое, чем «комплексные соединения».
Способность металлов к образованию координационных связей неодинакова.
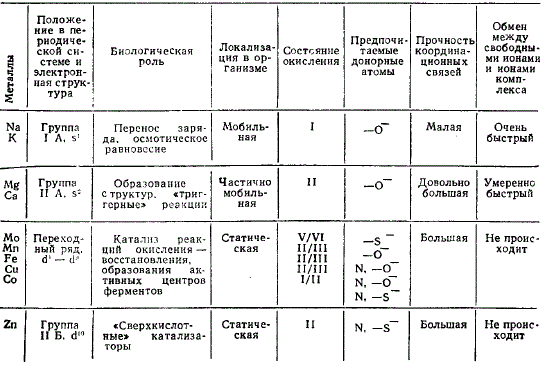
Биологическая роль металлов и их способность к комплексообразованию
В образовании координационных связей ионами металла главных подгрупп периодической системы (Na, К, Ca, Mg) важную роль играют электростатические взаимодействия; образующиеся комплексы весьма лабильны.
Переходные металлы (Mn, Fe, Co, Cu, Mo, Zn) дают координационные связи, имеющие преимущественно ковалентный характер. Они катализируют окислительно-восстановительные реакции и участвуют в образовании активных центров ферментов. Действие каждого металла специфично.
Наибольший интерес для биологов представляет, естественно, взаимодействие ионов металлов с ферментными системами. Это взаимодействие колеблется в широком диапазоне — от слабого ионного эффекта до образования комплексов.
Все ферменты, требующие для проявления максимальной активности присутствия металлов, можно разделить на две группы: 1) металлоферменты (или металлокоферменты) и 2) ферменты, активируемые металлом.
В металлоферментах металл является интегральным компонентом молекулы и не удаляется диализом. Извлечение его сильной кислотой или хелатирующим агентом приводит к снижению активности фермента. Ферменты этого типа обычно содержат в активном центре переходные металлы (Cu, Fe, Zn), формирующие очень стабильные координационные комплексы. Металл всегда присутствует в стехиометрических количествах, обычно от 1 до 4, иногда до 6—8 атомов на молекулу фермента (например, Fe в порфириновом комплексе каталазы, цитохромоксидазы). Некоторые из ферментов этого типа содержат два металла, например цитохромоксидаза (Fe и Cu), ксантиноксидаза (Fe и Mo).
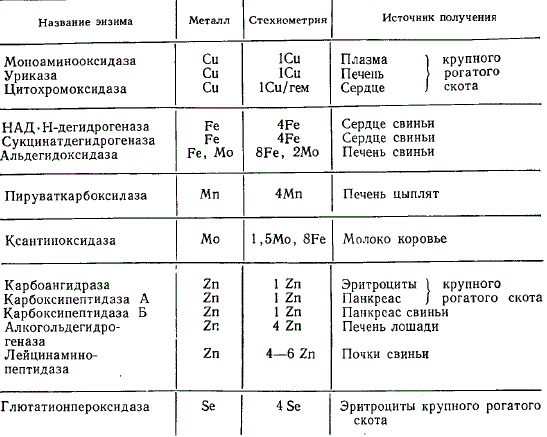
Некоторые металлоэнзимы организма животных
Потребность в металле у перечисленных ферментов не является специфичной во всех случаях без исключения. Так, цинк может быть извлечен из карбоксипептидазы А и замещен атомом кобальта. В пируваткарбоксилазе марганец может быть заменен магнием in vitro без потери активности.
Если в металлоферменте ион металла постоянно связан в составе активного центра, то, будучи прочной частью кофермента, ион может входить в активный центр и выходить из него. Примером может служить Co (III) в составе B12 — коферментов (соединений, родственных цианкобаламину). Эти соединения необходимы для активности металмалонил-КоА-мутазы и метионинсинтетазы (диолдегидразы).
В ферментативных реакциях, активируемых металлами, металл обычно не прочно связан с ферментом и может быть почти полностью извлечен диализом при pH 7,0. Стабильность такого комплекса невелика.
Следующие 15 катионов могут служить активаторами одного или нескольких ферментов: Na+, К+, Rb+, Cs+, Mg2+, Са2+, Zn2+, Cd2+, Cr2+, Cu2+, Mn2+, Fe2+, Co2+, Ni2+, Al2+.
Широко распространена взаимозаменяемость катионов, имеющих сходные ионные радиусы, электронную конфигурацию и электроотрицательность.
Например, ионы Mn2+, Fe2+, Co2+, Ni2+ активируют аргиназу in vitro. Все указанные четыре катиона относятся к первой переходной серии периодической системы. Различия в степени активации фермента этими ионами обусловлены лишь различиями в координационных комплексах, которые они образуют (электронной конфигурации и типе связей). А это, в свою очередь, обусловлено разной степенью насыщения 3d-орбиты у атомов данных элементов.
Каков же механизм влияния ионов металлов на каталитические свойства ферментов?
Прежде всего, ферменты, имея единую трехмерную структуру, первично обладают каталитической функцией. Металл требуется лишь для проявления ими максимальной активности.
Надо полагать, что конкретный механизм влияния иона металла на каталитическую активность будет зависеть от того, входит ли металл в структуру активного центра фермента или выполняет функцию активатора. В первом случае роль металла может заключаться:
а) в повышении селективности фермента по отношению к субстрату;
б) в непосредственном включении металла в катализ путем окисления и восстановления в ходе реакций, связанных с переносом электронов.
Не удивительно, что эти специфические функции может выполнять только определенный металл и только в определенном состоянии окисления (MnII/III, FeII/III, CuI/II, CoII/III и т. д.).
Во втором случае возможны следующие варианты каталитического влияния металла:
а) ион металла облегчает связывание субстрата с ферментом путем координации и изменения формы субстрата в соответствии со стерическими требованиями активного центра;
б) ион металла связывает одновременно кофермент и субстрат с ферментом;
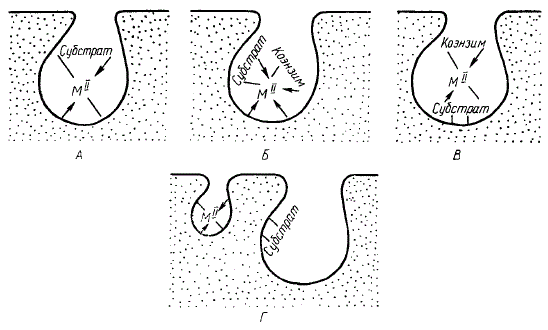
Функции металла в ферментных системах
в) металл вообще не связывается с ферментом, а образует комплекс с субстратом или коферментом, облегчая их соединение с активным центром;
г) ион металла связывает функциональные группы фермента, расположенные вне активного центра, но важные для стабилизации третичной и четвертичной структуры белка, пространственной конфигурации активного центра (например, влияние Ca++ на стабилизацию структуры α-амилазы);
Иногда в металлоэнзиме металл выполняет одновременно и каталитическую, и структурную функции (например, цинк в дегидрогенезах).
д) ион металла удаляет ингибитор, присутствующий в ферментном комплексе, или вытесняет малоэффективный ион из соединения с активным центром или функциональными группами субстрата.
В заключение отметим, что хелатные связи переходных металлов с нуклеиновыми кислотами (ДНК, РНК), наличие которых установлено за последние годы, возможно, окажутся биологически не менее важными, чем только что рассмотренные металлоферментные взаимодействия.